Risposta obiettiva completa e stabile da 5 anni col Metodo Di Bella di un carcinoma della mammella plurimetastatico dopo mastectomia, chemio e radioterapia.
Pubblicato il 04/01/2018
Risposta obiettiva completa e stabile da 5 anni col Metodo Di Bella di un carcinoma
della mammella plurimetastatico dopo mastectomia ,chemio e radioterapia
ABSTRACT:
Il cancro al seno è il tumore più comune ed è la principale causa di morte per cancro tra le donne. Nonostante tutti gli sforzi fatti, nel 2016 sono state stimate circa 11.939 morti e 50.000 nuove diagnosi di cancro al seno tra le donne italiane. Pertanto sono necessari nuovi approcci per migliorare la sopravvivenza e tassi più elevati di remissione. Presentiamo un caso di una donna con carcinoma della mammella e metastasi multiple dopo mastectomia destra, dissezione ascellare, cicli ripetuti di chemio e radioterapia e blocco degli estrogeni. Il metodo biologico formulato dal Prof. L. Di Bella (MDB) ha prodotto una risposta obiettiva completa e stabile senza tossicità. L' MDB comprende molecole antiproliferative, come la somatostatina, la prolattina e gli inibitori degli estrogeni insieme a molecole differenziative e apoptotiche come la melatonina (MLT), i retinoidi, la vitamina E, la D3, la vitamina.C, il calcio, amminozuccheri, associati a microdosi metronomiche di farmaci chemioterapici. Gli esami del sangue non hanno mostrato alcun danno ma una progressiva riduzione della prolattina, dell'estradiolo e dell'IGF1 e il mantenimento di bassi livelli di GH. Il risultato obiettivo di questo caso, in assenza di tossicità, dimostra l'efficacia del trattamento ed è in accordo con i risultati positivi già pubblicati sull'uso del MDB. Non richiedendo il ricovero ospedaliero o diurno e senza tossicità significativa, il MDB ha evitato i significativi effetti collaterali della chemioterapia e della radioterapia. Riteniamo che questo caso possa incoraggiare più interesse e studi più approfonditi sulle possibilità che sono state aperte in oncologia dal trattamento MDB del carcinoma mammario metastatico.
INTRODUZIONE:
Si stima che in Italia, nel 2016, ci siano stati 50000 nuovi casi di cancro al seno confermano che questo tipo di tumore è il più diagnosticato nella donna.
Nel 2013, il tumore al seno ha rappresentato la prima causa assoluta di morte in tutto il mondo. In Italia questo tumore ha causato 11.939 decessi (Fonte ISTAT 2016). Il cancro al seno presenta il 29% di cause morte prima dei 50 anni, il 21% tra 50 e 69 anni. e il 16% dopo 70 anni.
La sopravvivenza a 5 anni è dell'85,5% in Italia, un po 'più della percentuale Europea (84,7%). (I numeri del cancro in Italia, 2016)
Gli approcci attuali sono essere diversi e dipendono dal tipo di cancro. La procedura prevede chirurgia conservativa o radicale (mastectomia) associata a radioterapia e / o chemioterapia, che presentano anche variazioni a seconda del tipo e dello stadio del tumore (I numeri del cancro in Italia, 2016)
La stima in Italia risalente all'anno 2016, mostra 50.000 nuovi casi di cancro al seno che confermano che questo tipo di tumore è il più diagnosticato nella donna.
CASE REPORT:
Presentiamo il caso di una donna di 35 anni con carcinoma della mammella rilevato durante la gravidanza, conclusa alla 34 ° settimana da taglio cesareo a causa della diagnosi del carcinoma mammario. La biopsia mostrava che si trattava di un carcinoma infiltrante (G2.
La paziente è stata sottoposta a mastectomia destra con dissezione ascellare e ricostruzione plastica il 3.12.2009. L’esame istologico relativo riporta la seguente diagnosi: “Carcinoma duttale a medio grado di differenziazione, con focale componente intraduttale di tipo solido e con estesa invasione vascolare peritumorale. Parenchima mammario esente da neoplasia, con modificazioni di tipo gravidico. Focolai di neoplasia intraepiteliale dei dotti (DIN2) con estensione ai dotti retroareolari ed estesi fino in prossimità del margine retroareolare. Metastasi linfo-perilinfonodali di carcinoma a due linfonodi sentinella e ad un linfonodo di II livello. Istiocitosi dei seni nei rimanenti ventiquattro linfonodi esaminati”, estadiazione pT2(2.5cm) pN1a(3/27) MX G2. Invasione vascolare estesa; ER 90%, PgR 10%, Ki67 15%”, c-erbB2: debole completa nel 70%.
La paziente è stata sottoposta a 4 cicli di chemioterapia con protocollo AC e terapia ormonale con Decapeptyl (3,75 mg / mese) + Tamoxifene (da 20 mg die).
Una PET / TC eseguita il 10 maggio 2010 rileva una captazione splenica, confermata da una successiva ecografia addominale che mostra una piccola area ipoecogena di circa 8 mm in corrispondenza della parte superiore della milza.
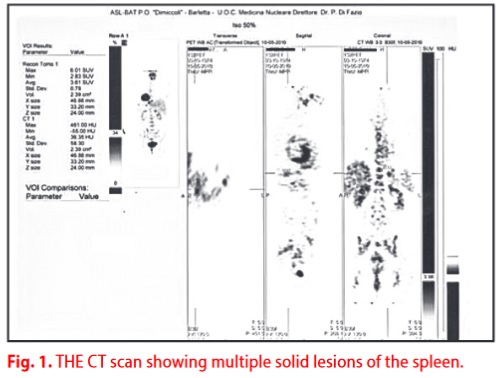
La TC eseguita il 26 maggio 2010 ha mostrato multiple lesioni focali spleniche solide di natura ripetitiva(Figura 1).
Nell'aprile 2011, il paziente è stato sottoposto a sostituzione dell'estensore mammario con protesi e mastoplastica riduttiva sinistra.
Alla RM di controllo nel maggio 2011, non è stata mostrata alcuna progressione delle lesioni spleniche, tuttavia l'ecografia e la mammografia hanno mostrato una retrazione, probabilmente chirurgica, sul lato destro che necessitava di ulteriori indagini. L'ecografia addominale eseguita a novembre risultava negativa.
Le analisi del sangue eseguite ad aprile 2012 hanno mostrato un incremento del marcatore tumorale Ca 15.3 pari a 35,6 per cui la paziente inizia una serie di accertamenti con specifici esami strumentali. La PET ha rivelato iperaccumuli patologici ai tessuti molli della regione mammaria sinistra, ai linfonodi parasternali, ai linfonodi dell’ilo epatico, a livello epatico ed al rachide lombare.
La TC e la RM hanno rivelato l'adenopatia ascellare destra, l'adenopatia subdiforme e sub-diaframmatica e lesioni ossee nell'ileo destro.
Nel giugno del 2012 la paziente inizia il Metodo Di Bella.
La prescrizione comprendeva:
Ac Retinoico gr 0,5;
axeroftolo palmitato gr 0,5;
betacarotene gr 2 in alfa tocoferile acetato gr 1000 un cucchiaio mattino ,mezzodì , e sera almeno 15’ prima del pasto, aggiungendo nel cucchiaio Diidrotachisterolo 12 gocce nel cucchiaio per somministrazione (36/dì)
Decapeptyl 3,75 mg una fiala intramuscolo ogni 4 settimane ;
Somatostatina fiale da 1 mg. Aspirare una fiala (da 1mg ) di somatostatina in una siringa da 10 ml e aggiungere soluzione fisiologica fino riempire completamente tutta la siringa .Collegare la siringa al temporizzatore, innestare nella siringa un ago Micro-flo T GA 27 da 0,4mm x10 mm e iniettare sottocute la sera 3 ore dopo cena regolando il temporizzatore a 12 ore. Almeno per il 1° mese, poi a 10 ore) Iniziare con una sola fiala (1 mg ), (riempiendo tutta il resto della siringa confisiologica) aumentando di una fiala (1 mg) la settimana fino a tre fiale (3 mg );
Tetracosactide 0,25 mg fiale nella stessa siringa con la somatostatina a giorni alterni compatibilmente con pressione e glicemia;
Octreotide a lento rilascio 20 mg ogni 20 giorni;
MelatoninaMDB 5 mg ,tre compresse mezzodì ,e la sera ai pasti , e 10 prima di coricarsi ( solubili in acqua ) 16 cpr al dì per complessivi 80 mg;
La paziente assumeva anche i seguenti farmaci:
Dostinex: mezza compressa a mezzodì 2 volte la settimana;
Parlodel 2,5 mg mezza compressa mattino e sera;
Arimidex una cpr al dì(in sostituzione del Tamoxifene 1 cp die);
Endoxan 50 mguna cpr mattino e sera;
Ac Ascorbico (Vit C) ½ cucchiaino in un bicchiere d’acqua mezzodì e sera durante il pasto con
Calcium Sandoz 1\2 bustina nello stesso bicchiere;
Condroitinsolfato 500 mg 2 cps mattino e sera;
Calciolevofolinato 22 mg una cps al giorno;
Zofran Se vomito una compressa da 8 mg ,o una supposta o una fiala intramuscolo da 8 mg;
La paziente è stata sottoposta a biopsia delle lesioni ossee del'ileo che ha confermato una massiccia metastasi da carcinoma mammario (ER 40%, PgR 10%, Her2 debole).
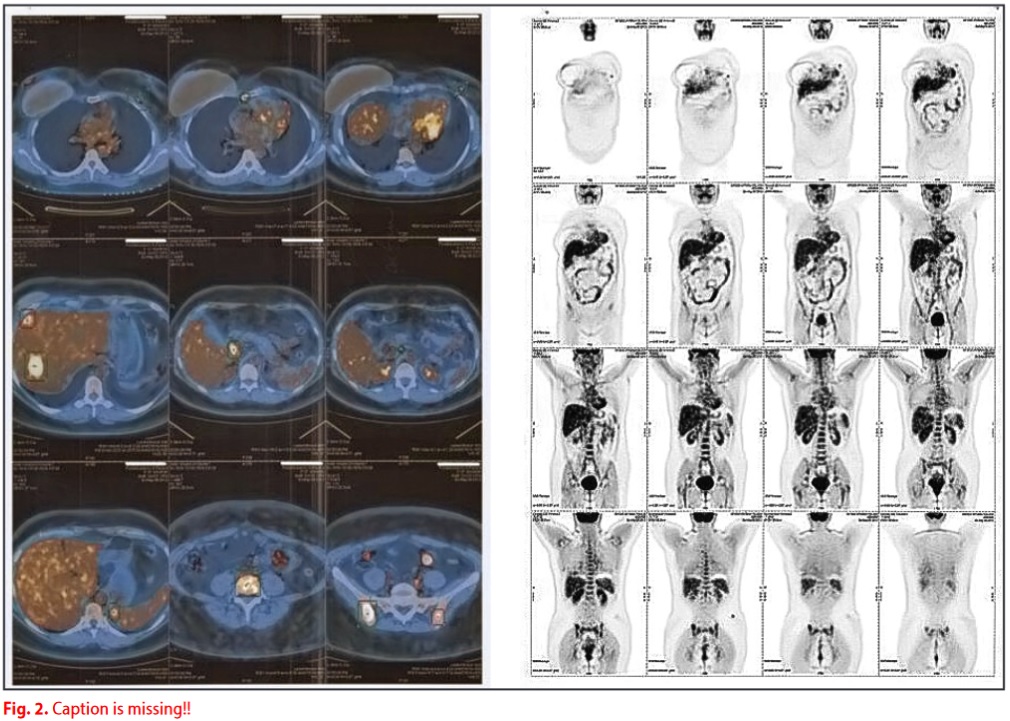
Una revisione dei vetrini per la valutazione della cromo-granina ha mostrato positività nel 20% delle cellule tumorali. Dopo sei mesi di trattamento con DBM, la PET ha mostrato una regressione più o meno completa dell'assorbimento epatico, osseo e linfonodale.
Una PET eseguita dopo sei mesi di applicazione del MDB evidenzia una regressione pressoché completa delle captazioni a livello epatico, osseo e linfonodale.
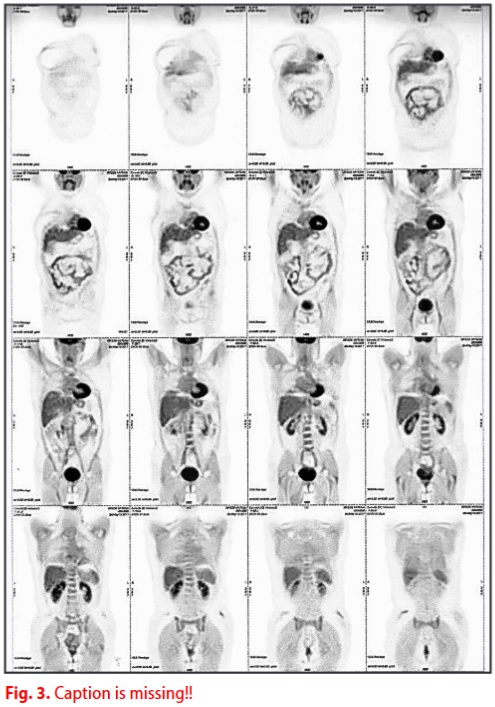
Nella successiva PET di controllo eseguita nel gennaio 2014, non sono stati riscontrati accumuli anomali (ad eccezione di alterazioni spleniche aspecifiche). Nel gennaio 2014 il paziente ha sospeso anche la terapia con Endoxan e continuato con le terapie ormonali / biologiche.
Successivi test strumentali hanno mostrato la completa remissione della malattia.
Da gennaio 2014, il paziente ha subito diverse scansioni PET di tutto il corpo, l'ultima nel maggio 2017, tutte senza recidiva della malattia.
LA TERAPIA E IL DECORSO CLINICO
La paziente, nel giugno del 2012 , per la ripresa di malattia, dopo aver rifiutato un secondo ciclo di Chemioterapia che le era stato proposto, ha chiesto di essere curata con il Metodo Di Bella, che prevede l’impiego sinergico di molecole ad azione differenziante, citostatica, apoptotica, antiproliferativa, con incremento dell’attività immunitaria. La somministrazione continuativa per circa 6 mesi di 100 milligrammi di ciclofosfamide al giorno, insieme a tutti i componenti del MDB, per l’azione mieloprotettiva, antidegenerativa e trofica su parenchimi e tessuti soprattutto della MLT e degli alti dosaggi di Vitamina E, Retinoidi, vitamine C e D3, non ha causato alcuna tossicità midollare, epatorenale, metabolica, cardiocircolatoria, neurologica, né depressione immunitaria. Non ha provocato rilevanti alterazioni della crasi ematica e della dinamica midollare. Nella terapia Di Bella viene utilizzato Endoxan (ciclofosfamide) in dosi che oscillano tra 50 e 100 milligrammi al giorno con finalità apoptptiche , non citolitiche come nei protocolli oncologici . Confrontando 100 mg /die di ciclofosfamide del MDB con i 10 ai 12 grammi in vena nelle monoterapie pretrapianto delle malattie linfoproliferative si ha un rapporto di 1 a 100. 100 milligrammi, invece di 10 grammi, non si ottiene un effetto citolitico o citotossico, ma un effetto totalmente diverso, apoptotico. Nel Nel giro di sei mesi la paziente, conducendo a domicilio la terapia ha ottenuto una remissione parziale prima, e completa subito dopo, che le ha permesso di tornare al lavoro.
DISCUSSIONE E RAZIONALE DELLA TERAPIA
La perdita della differenziazione e della proliferazione, anche se in misura diversa, sono denominatori comuni di tutte le neoplasie. L'espressione ubiquitaria del recettore della prolattina (Ben-Jonathan et al., 2002; Hooghe et al., 1998) e del GH (Lincoln et al., 1998; De Souza et al., 1974) sono una delle conferme del ruolo mitogenico diretto e generalizzato di questa molecola. La proliferazione cellulare è altamente dipendente dalla prolattina e dal GH, entrambi potenti fattori di crescita e da molecole mitogene dipendenti dal GH che sono regolate positivamente da esso, come EGF, FGF, HGF, IGF1-2, NGF, PDGF, VEGF e TGF in aggiunta a fattori di crescita prodotti dal tratto gastrointestinale, come VIP, CCK e PG. Sia la proliferazione cellulare fisiologica che quella neoplastica avvengono per mezzo di queste stesse molecole, che le cellule neoplastiche utilizzano però in misura esponenziale rispetto a quelle sane.
Gli antidoti biologici al GH come la somatostatina e composti simili riducono non solo l'espressione e la trascrizione di fattori di crescita altamente mitogenici, come IGF1-2 (Cascinu et al., 2001; Pollak 1997; Schally et al., 2001), EGF (Held- Feindt et al., 1999) e FGF (Mentleinet al., 2001), ma estendono la loro regolazione negativa ai rispettivi recettori con evidenti effetti anti-proliferativi e anti-angiogenici (Szepesházi et al., 1999, Mishimaet al., 1999). L'estensione dell'asse GH-IGF1 sullo sviluppo biologico neoplastico è degna di nota. Gli IGFR rispondono mitogenicamente all'IGF. L'effetto suppressivo dell'SST sui livelli sierici di IGF1, è sia diretto, inibendo il gene IGF1, sia indiretto sopprimendo GH e quindi la sua induzione epatica di IGF1.
L'angiogenesi è essenziale per la progressione neoplastica. L'angiogenesi è a sua volta regolata dalla cascata di monociti, interleuchina 8 e da fattori di crescita come VEGF, TGF, IGF1, FGF, HGF e PDGF. Ognuno di questi fattori è regolato negativamente dalla somatostatina e dal suoi omologhi sintetici (Albini et al., 1999; Barrie et al., 1993; Cascinu et al., 2001; Florio et al., 2003; Jia et al., 2003; Turner et al., 2000 ; Vidal et al., 2000; Watson et al., 2001; Wiedermann et al., 1993). L'inibizione dell'angiogenesi indotta da SST è potenziata sinergicamente dall'MLT (Lissoni et al., 2001 Di Bella et al., 1979; Di Bella & Gualano 2006), dai retinoidi (McMillan et al., 1999; Kini et al., 2001; Majewski; et al., 1994), dalla vitamina D3 (Kisker et al., 2003; Mantell et al., 2000), dalla vitamina E (Shklar & Schwartz 1996; Tang & Meydani 2001; Neuzil et al., 2002), vitamina C (Ashino et al., 2003),dagli inibitori della prolattina (Turner et al., 2000) e dai componenti della matrice extracellulare (Liuet al 2005; Ozerdem & Stallcup 2004).
Allo stesso modo l'effetto citostatico, anti-proliferativo e anti-metastatico della somatostatina è efficacemente in sinergia con gli altri componenti di MDB:
Retinoidi (Hassan 1990; Voigt et al., 2000; Piedrafita & Pfahl 1997; Onogi et al., 1998);
MLT (Bartsch et al., 1999; Kvetno & Levin 1986; Mediavilla et al., 1999; Maestroni et al., 1996; Cos et al., 2000);
Vitamina D3 (Jensen et al., 2001; Barroga et al., 2000; Campbell et al. 2000);
Cabergolina e bromocriptina (inibitori della prolattina) (Gruszka et al., 2001; Ben-Jonathan et al., 2002; Lissoni et al., 2001; Klijn et al., 1996; Manni et al., 1989);
Glucosamina solfato, galattosamina solfato, componenti della matrice extracellulare (Pumphreyet al 2002; Batra et al., 1997);
Vitamina E (Turley et al., 1995; Israel et al., 2000; Malafaet al 2002; Neuzil et al., 2002; Shklar & Schwartz 1996);
Vitamina C (Head 1998; Murata et al., 1982; Cameronet al. 1979).
La relazione causale tra l'espressione del recettore del GH e l'induzione della progressione tumorale è stato dimostrata (Lincol 1998), dimostrando istochimicamente concentrazioni significativamente più elevate di GHR nei tessuti tumorali rispetto ai tessuti fisiologici, mostrando così il potente ruolo mitogenico del GH con indici proliferativi a seconda della dose. Questo è diretto, tramite recettori, oltre che indiretto, inducendo l'espressione epatica di IGF1, che è dipendente dal GH. L'asse GH-IGF1 ha un ruolo decisivo nel comportamento biologico di molte neoplasie. In un'alta percentuale di cellule neoplastiche, sono stati identificati i recettori IGF1 che rispondono mitogenicamente al ligando.
La somatostatina esercita un effetto antiblastico sia direttamente, inibendo l'espressione del gene IGF1, sia indirettamente, sopprimendo GH, che è necessaria per l'incretione di IGF1 (Pollak 1997; Schally et al., 2001; Schally & Nagy 2003). L'attività inibitoria dell'SST sull'EGF, un altro potente fattore di crescita mitogenico, con meccanismi multipli, è stata inoltre accuratamente documentata:
- in base al dosaggio, inibizione della fosforilazione della tirosina indotta dall'attivazione di EGFR da parte di EGF (Mishima et al., 1999);
- riduzione dell'EGFR nelle cellule tumorali (Szepesházi et al., 1999);
- riduzione dell'espressione di EGF (Held-Feindt et al., 1999);
- riduzione della concentrazione plasmatica di EGF (Cascinuet al., 2001).
Gli ormoni mitogeni prodotti dal tratto gastrointestinale come VIP, CCK e PG sono fortemente inibiti dalla somatostatina e/o dall'octreotide (Kath & Höffken 2000), ed è stato dimostrato che i tumori al seno esprimono i recettori SSTR1, SSTR2 e SSTR3 e meno frequentemente SSTR5 (Albérini et al., 2000; Schaer et al., 1997). Pertanto la presenza di SSTR (Barnett 2003, Pinzani et al., 2001, van Eijck et al., 1998) e di recettori neuroendocrini in una percentuale significativa di questi carcinomi costituisce un'ulteriore indicazione razionale per l'uso dell'SST, che in ogni caso è già stato ampiamente giustificato dall'effetto negativo sopra descritto di SST su GH, oncogeni e angiogenesi correlati a GH.
Angiogenesi e neoangiogenesi, sono condizioni necessarie per la progressione del tumore, così come la cascata di monociti, il rilascio di interleuchina 8 (IL-8) e il contributo di GFs (il cui sinergismo è essenziale), sono bersagli molecolari specifici regolati negativamente dalla somatostatina e dai suoi analoghi (Ruscica et al., 2012). L'inibizione dell'angiogenesi indotta da SST è rinforzata in modo sinergico da MLT, retinoidi e vitamina D3 (Kim et al 2013, Lissoni et al., 2001 Sogno et al., 2009 Picotto et al., 2012). Inoltre, le condizioni locali di ipossia/anossia e acidosi promuovono l'angiogenesi e sono per lo più corrette dal miglioramento degli scambi ematici indotti dai componenti differenzianti del MDB. Allo stesso tempo, gli effetti citostatici, antiproliferativi e antimetastatici della somatostatina sono aumentati sinergicamente dagli altri componenti del MDB. Un ulteriore contributo è fornito dalla somministrazione giornaliera di basse dosi (50-100 mg / die per os) di Ciclofosfamide (Endoxan®). Oltre a ridurre drasticamente gli effetti anitblastici/mielosoppressivi noti, questo dosaggio induce una marcata inversione dei suoi meccanismi d'azione quali:
innesco della cascata apoptotica dipendente dai mitocondri;
azione anti-angiogenetica mediante drasticamente down-regolazione dell'espressione del gene VEGF (Loven et al. 2013).
Numerose indagini precliniche hanno anche dimostrato i meccanismi di azione della MLT. L'uso di tale indolo si estende a tutti gli istotipi del cancro al seno a causa della sua alta distribuzione recettoriale/nucleare (Oprea-Ilies et al., 2012; Rögelsperger et al., 2011). Poiché la molecola è associata alle vie di segnalazione dello sviluppo epiteliale sia fisiologico che neoplastico, questa sostanza ha le proprietà di neutralizzare selettivamente i segnali proliferativi degli estrogeni e di modulare negativamente la loro biosintesi locale (Hill et al., 2011; Girgert et al. 2009).
La somministrazione di basse dosi di inibitori dell'aromatasi di seconda generazione (Anastrozole ©), già utilizzata nella pratica clinica, combinata con MLT, SST e Retinoidi, regola negativamente i processi ormonali di proliferazione dei tumori della mammella (Alvarez-Garcia et al. 2013; Margheri et al., 2012; Wang et al., 2012; Knower et al., 2012; Ciolinoet al., 2011).
Bibliografia:
Barnett P (2003). Somatostatin and somatostatin receptor physi-ology. Endocrine. 20(3): 255–264.
Kvetno IM, Levin IM (1986). Melatonin and tumor growth. (In Russian with English abstract). Eksp Onkol. 8(4): 11–15.